WrigleysHaze
Member
Hi,
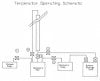
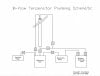
I have a 2 lbs closed loop extractor active (but it only fits max 1.5 lbs) and I have a few basic questions and problems. Unfortunately, I am currently working without pressure support, but this should change in 1 - 2 weeks.
My biggest problem is that after rinsing, when there is already more than approx. 70% in the collecting tank, the rest of the solution is still in the material chamber and keeps running out in small waves. I could understand constant running or dripping, but there are always a few ml and then a short pause and then something comes out again, etc.
That's why I usually flush 2-3 times and add about 13.5-18 lbs per flush. 100% propane @ -22°F to -40°F.
I get ~50g on the first wash, ~15-20g on the second and 5-10g on the third.
Is this due to the lack of pressure support or am I using too little solution or is it normal that you can/should wash several times?
and finally, could someone tell me something about my product?

In Collecting chamber

After scratching together

After 48h in vacuum at ~100°F
What did I do there and why is it so different, should I mix it?
I have a 2 lbs closed loop extractor active (but it only fits max 1.5 lbs) and I have a few basic questions and problems. Unfortunately, I am currently working without pressure support, but this should change in 1 - 2 weeks.
My biggest problem is that after rinsing, when there is already more than approx. 70% in the collecting tank, the rest of the solution is still in the material chamber and keeps running out in small waves. I could understand constant running or dripping, but there are always a few ml and then a short pause and then something comes out again, etc.
That's why I usually flush 2-3 times and add about 13.5-18 lbs per flush. 100% propane @ -22°F to -40°F.
I get ~50g on the first wash, ~15-20g on the second and 5-10g on the third.
Is this due to the lack of pressure support or am I using too little solution or is it normal that you can/should wash several times?
and finally, could someone tell me something about my product?
In Collecting chamber
After scratching together
After 48h in vacuum at ~100°F
What did I do there and why is it so different, should I mix it?